说明:本文以轨道杂化为核心,系统揭示s-p-d-f多轨道耦合与自旋构型如何精准调控吸附强度、电子转移与反应路径;读者可据此把握从原子轨道层面设计高效催化剂的普适策略,快速锁定提升活性、选择性与稳定性的关键杠杆,洞察实验现象背后的电子本质。
轨道杂化的重要性
催化剂活性表面的电子结构对催化性能具有重要影响,因此电子结构常被用作评估设计催化剂优劣的有效且精准的指标。本质上,电子结构可视为相关原子轨道的线性组合,它决定了电子的行为与性质。
通过调控催化活性位点轨道(简称“活性轨道”)的取向或改变其自旋态,可优化催化剂对吸附质的吸附能力,或满足催化反应步骤中有利的自旋选择规则。
轨道分裂、占据情况、取向及自旋态等常见轨道特征相互影响,且对配位配体场等周围环境高度敏感。在催化剂内部的轨道杂化过程中,这些特征会共同作用,使活性轨道与吸附质(反应物)形成具有特定活性、选择性甚至稳定性的相互作用。
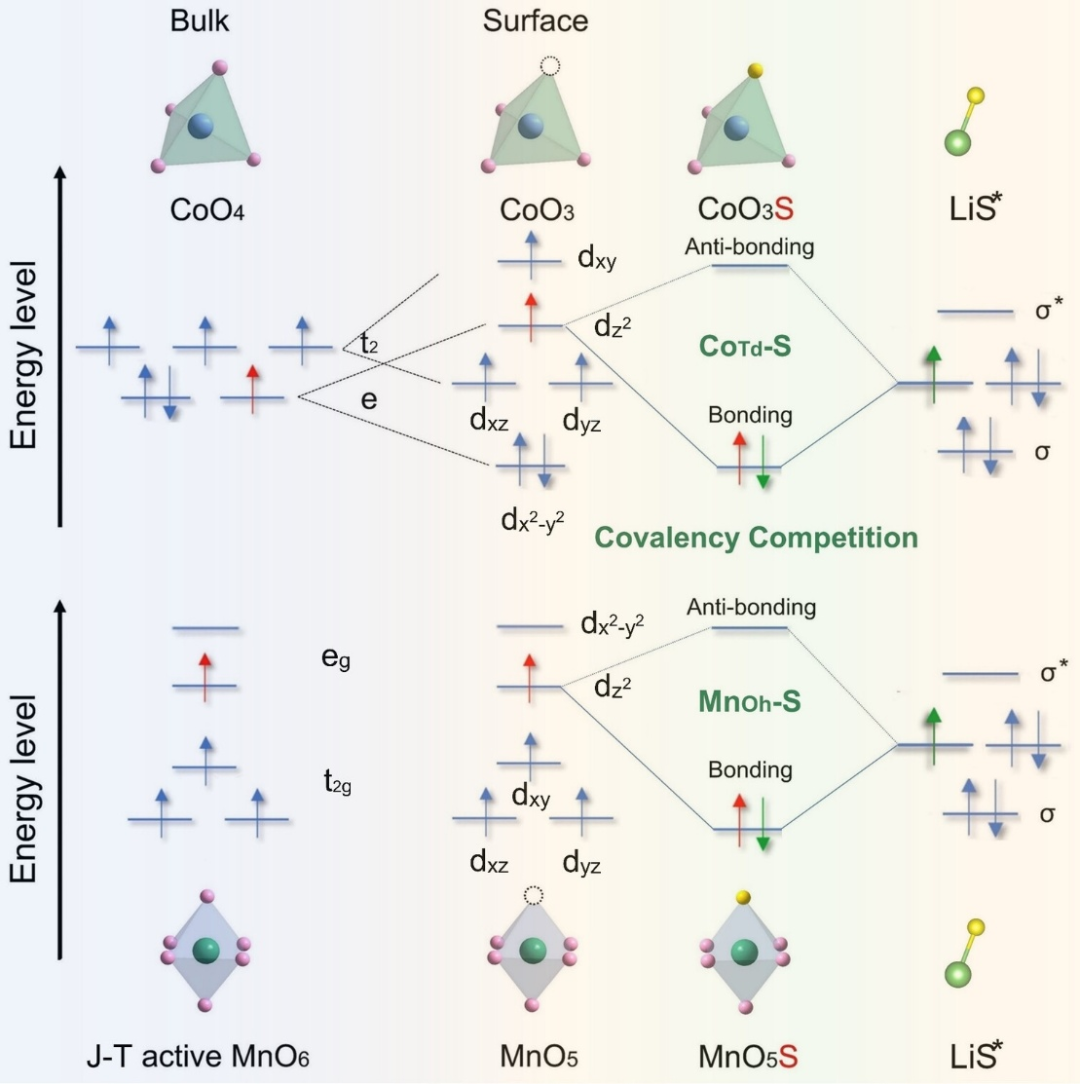
图1. 示意图:LiS*中间体与Co³⁺Td和Mn³⁺Oh位点的3d轨道之间的有利相互作用。DOI: 10.1002/anie.202216286
轨道杂化主要分为两类:一类从催化过程角度出发,聚焦催化剂与生成中间体之间的直接轨道耦合;另一类聚焦催化剂内部的轨道相互作用,以实现更有利的直接轨道耦合。
这两类杂化本质上分别通过调控反应过程与催化剂本身(如优化能量匹配、调节电子填充态、实现适度轨道重叠、满足有利自旋选择、调控构型转变),达到热力学与动力学均有利的催化坐标。
对于大多数吸附质而言,s轨道和p轨道是其主要功能轨道,当它们与丰富的活性位点相互作用时,会通过s (p)-s (p, d, f) 耦合形成多种相互作用。
s–s、s–p与s–d轨道相互作用
孤立原子的s轨道呈球形对称,与其他轨道耦合时空间相互作用概率均一,且易形成σ键。以典型的H原子为例,位点-H键强度常被用作评估光催化析氢、电催化析氢及多相加氢等涉氢反应的指标。
(1)s-s轨道相互作用的典型场景是小分子异裂解离。缺陷氧化铟(In₂O3-x(OH)y)是典型的受阻路易斯对(FLP)材料,其表面可通过s-s轨道耦合实现H₂的异裂解离。
(2)s-p轨道相互作用主要发生在主族元素(如P、S)活性位点与H/小分子吸附质之间,核心是通过s(如H1s)与p(如P 2p、S 3p)轨道的适度重叠,优化吸附强度与反应动力学。
如图2所示,研究人员采用阴离子取代策略,设计出自立式面NiP₂/Ni₅P₄异质结构。理论计算结果表明,H更倾向于吸附在P位点上,且该位点的ΔGH*(氢吸附自由能)比邻近Ni位点更负。
此外,H的1s轨道与P的2p轨道之间形成适度的轨道重叠,使催化剂具有适宜的氢吸附强度,在酸性条件下,电流密度达到100mA cm⁻²时过电势仅为76mV。
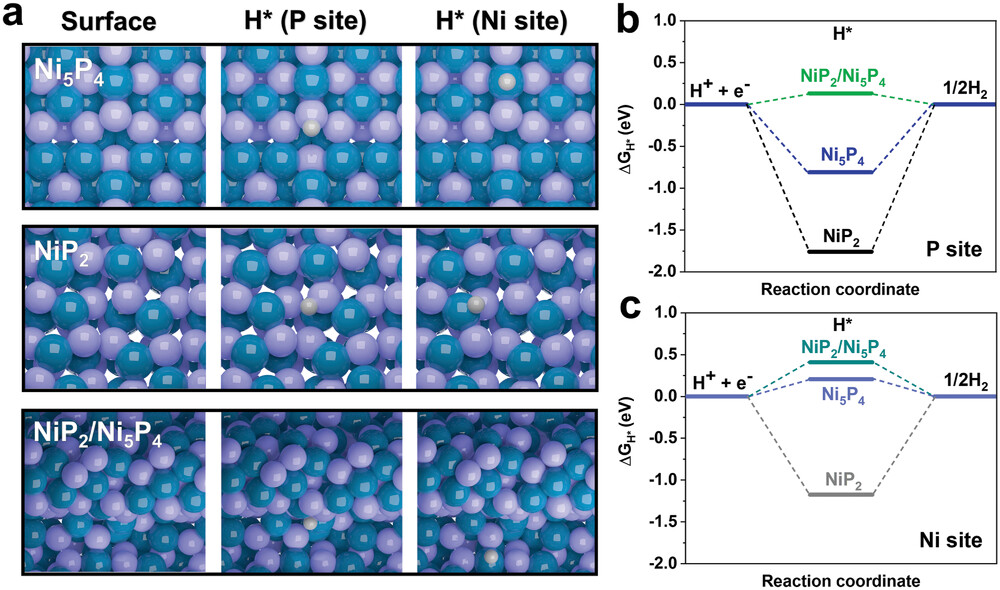
图2. (a)Ni₅P₄、NiP₂及NiP₂/Ni₅P₄模型的表面、H在P位点的吸附结构以及H在Ni位点的吸附结构。深绿色:Ni;浅蓝色:P;灰色:H。(b)在零电位下,催化表面P位点的析氢反应自由能图。(c)在零电位下,催化表面Ni位点的析氢反应自由能图。DOI: 10.1002/smll.202105642
(3)s-d轨道相互作用,过渡金属活性位点的核心耦合机制。过渡金属(TM)因费米能级附近存在局域锐化d轨道,其s-d轨道相互作用(H 1s与TM d轨道)是调控涉氢反应的关键。
通过引入空位可精细调控活性位点的电子态与配位态。基于这一思路,如图3所示,研究人员提出了一种空位介导的轨道导向策略,设计用于HER的催化剂。
通过氢气氛围退火构建N空位,模拟显示适度N空位使H在N/Fe位点的吸附能分别为-0.08/-0.14eV(接近热力学中性);进一步分析发现,N空位不足/过量时,N/Fe的空轨道呈垂直表面取向(虽利于H吸附,但阻碍脱附),而适度空位使N轨道倾斜、Fe轨道功能改变,电子耦合从H 1s-N pz转为H 1s-Fe d,同时优化H吸附与脱附。
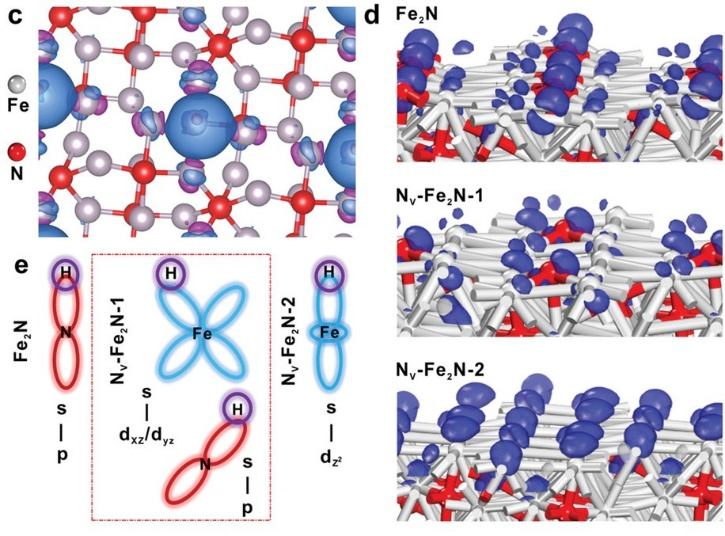
图3.(c)NV-Fe₂N-1表面N空位周围的电荷密度差图,蓝色表示电子流出,紫色表示电子流入。(d)Fe₂N、NV-Fe₂N-1和NV-Fe₂N-2在费米能级附近的未占据轨道分布。(e)不同取向下的轨道相互作用示意图。DOI:10.1002/adma.201904346
p-p与p-d轨道相互作用
与s轨道不同,p轨道具有三个简并轨道(pₓ、py、pz),呈纺锤形,且相互之间取向不同。
(1)过渡金属的d轨道具有局域性,当轨道能级匹配时,其与小分子发生相互作用的概率较高。而主族金属(如s区或p区金属)通常具有闭合d壳层或离域s/p能带,催化活性较低。通过调控p-p轨道相互作用,可使催化剂获得适度的吸附/脱附能力、更高效的电子转移能力,并降低催化过程中关键中间体的能垒。
如图4所示,研究人员发现,设计的褶皱超薄Rh₂P纳米片可作为全pH范围下高活性、高稳定性的HER催化剂。其关键在于引入了P的3p轨道:P的3p轨道不仅能与Rh的4d轨道通过s-d相互作用促进质子-电子电荷交换,还能增强p轨道重叠,使O相关物种(决速步涉及的物种)处于有利的吸附位置。
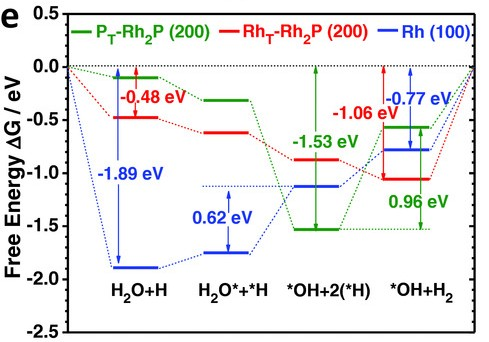
图4. (e)在碱性条件下,P端基表面(PT-Rh₂P (200))、Rh端基表面(RhT-Rh₂P (200))和Rh (100)表面析氢反应的自由能路径(ΔG)。DOI: 10.1002/aenm.201801891
(2)p-d轨道相互作用通过过渡金属d轨道与主族元素p轨道的能级匹配与电子耦合,实现活性位点电子态、中间体结合能的调控,是更丰富的相互作用模式。
如图5所示,单原子Nb在几何优化后略微突出于基底平面,Nb-N位点的局域畸变导致Nb d轨道分裂(dxz/dyz、dz²能级升高,dxy、dx2₋y2能级降低)。Nb-N₄单元产生的充足反键轨道,通过捕获-耦合-转化机制增强Nb d轨道与锂多硫化物(LiPSs)中S p轨道的p-d杂化,实现LiPSs的高效固定与催化转化。
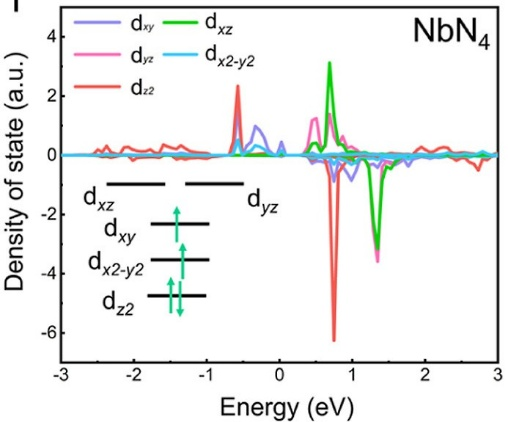
图5. Nb-d轨道的态密度(PDOS)。DOI: 10.1021/jacs.2c10345
d–d与d–f轨道相互作用
d轨道可容纳更多电子,具有五个简并轨道,且对周围环境高度敏感。这一特征使得d轨道在能级重分布、简并度消除、电子重新分布与填充以及自旋变化等方面具有丰富的多样性。
(1)d-d轨道相互作用通过TM间d轨道的能级匹配与电子耦合,优化电子转移速率、中间体结合能及反应路径。
如图6所示,研究表明,与面心立方(FCC)结构的PdCu纳米颗粒相比,体心立方(BCC)结构的有序PdCu纳米颗粒是更高效的氨合成电催化剂。在BCC结构PdCu中,Cu的3d轨道能级与Pd位点的两个主峰能级高度匹配,有效消除了eg-t2g分裂能隙,意味着原位库仑势垒更低,电子转移速率更高。
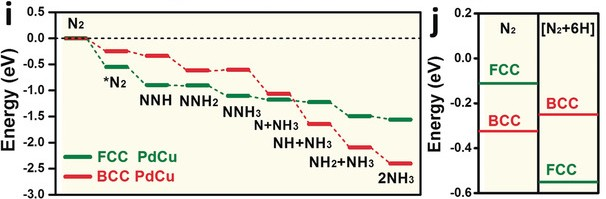
图6.(i)氮还原反应(NRR)在体心立方(BCC)PdCu和面心立方(FCC)PdCu 表面上的能量路径对比。(j)N₂+6 H与N₂分别直接吸附于体心立方(BCC)PdCu 和面心立方(FCC)PdCu表面的吸附行为比较。DOI: 10.1002/anie.201913122
(2)过渡金属与镧系金属之间的d-f耦合比过渡金属-过渡金属(TM-TM)耦合更具优势,因为前者的轨道排斥力更小。此外,催化剂中的f轨道还可引发5d-4f或5d6s-4f电子相互作用,有利于小分子活化。
实际上,过渡金属之间可通过直接成键形成d-d轨道相互作用,但考虑到排斥力的存在,部分d-d相互作用需通过桥连非金属元素实现,形成d-p-d电子构型。
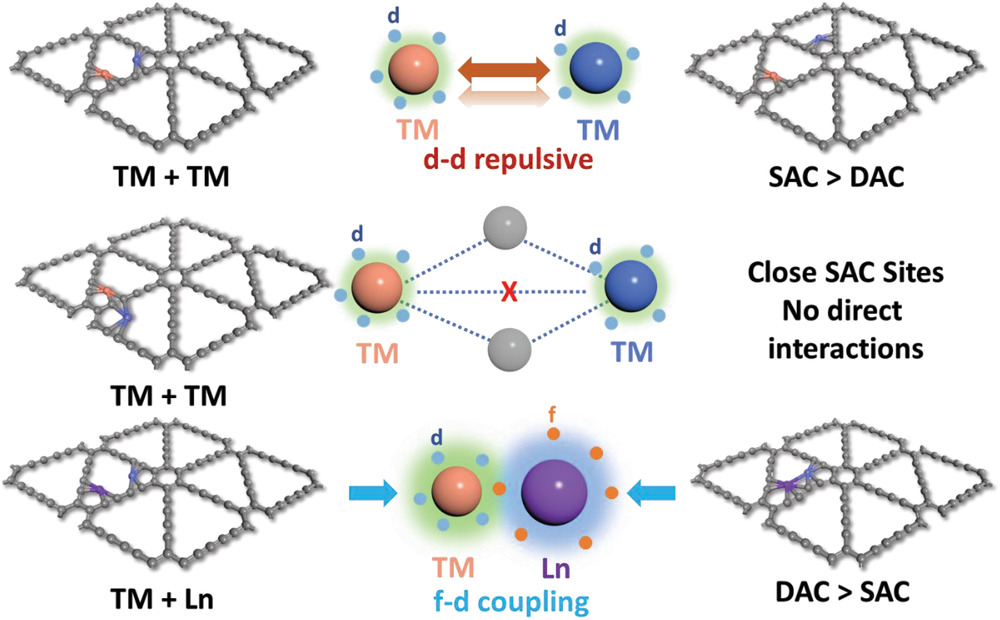
图7. 双原子催化剂(DAC)发展示意图。不同金属的组合会带来差异化的相互作用,并最终生成各异的合成产物。DOI: 10.1002/aenm.202003796
多轨道相互作用
轨道杂化对催化过程和催化剂的调控主要分为两类。当经过调控的催化剂投入实际应用时,会出现多轨道耦合现象,从整体视角来看,上述总结的大多数轨道相互作用均属于这一范畴,例如s-p-d、d-p-d及d-p-f轨道耦合。
如图8所示,研究人员开发了一种多孔氮掺杂石墨烯负载的Fe/Cu双原子催化剂,用于硝酸盐还原反应。研究发现,Fe向Cu的电子转移导致金属-金属二聚体产生极化,该极化二聚体对硝酸盐还原过程中的大多数中间体均表现出适度的结合能。
因此,双原子中心与NO₃⁻之间形成强相互作用:更准确地说,Fe/Cu的3dxz轨道与NO₃⁻中两个氧原子的2px轨道发生耦合。
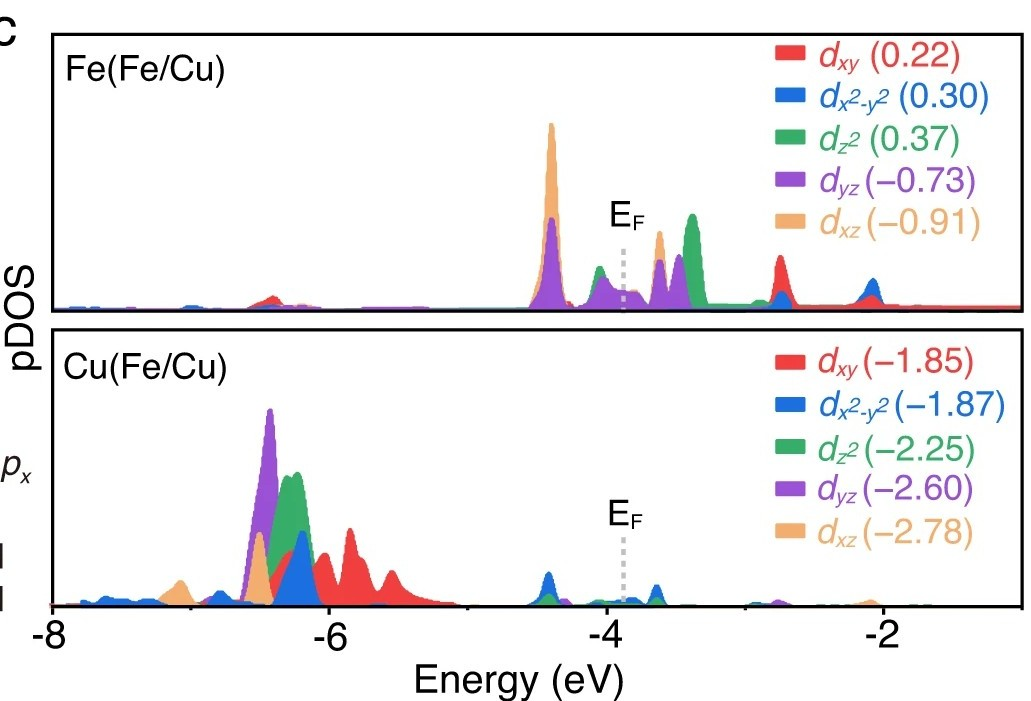
图8. (c)Fe/Cu 3d轨道的投影态密度(PDOS)。(d)NO₃⁻在Fe/Cu-HNG表面上相互作用的Wannier轨道。DOI: 10.1038/s41467-023-39366-9
轨道的自旋构型
轨道杂化不仅涉及电荷自由度,还与自旋自由度密切相关,这对高效催化剂的预测具有重要意义。
自旋态与电子结构、轨道分裂及分布密切相关,且可根据晶体场分裂能与自旋配对能的相对大小发生重排。当晶体场分裂能小于自旋配对能时,易形成高自旋态。高自旋电子或新形成的自旋态对吸附质轨道敏感,有利于特定自旋演化/维持过程,从而促进吸附与活化。
如图9所示,ORR过程中单原子Fe单元捕获*O₂⁻和*OH⁻中间体的具体作用。不同场景下演化的中间体揭示了D3单元的结构与动态循环过程,其中自旋构型发生显著变化,具体结构与动态循环如图9所示。
FeⅡN₄C₁₂的D1结构产生低自旋Fe²⁺,而 FeⅡN₄C₁₀的D2结构与N-FeⅡN₄C₁₀的D3结构分别产生中自旋与高自旋Fe²⁺。在0.9 V电势下,O₂吸附于D3位点,Fe的3d轨道与O的反键π轨道发生杂化,生成 O₂⁻-FeⅡN₅中间体(操作型拉曼光谱证实),同时四极分裂与同质异能移值显著降低。
这些结果表明,中心 Fe²⁺从高自旋态急剧转变为低自旋态,且结构向N₄平面结构演化。
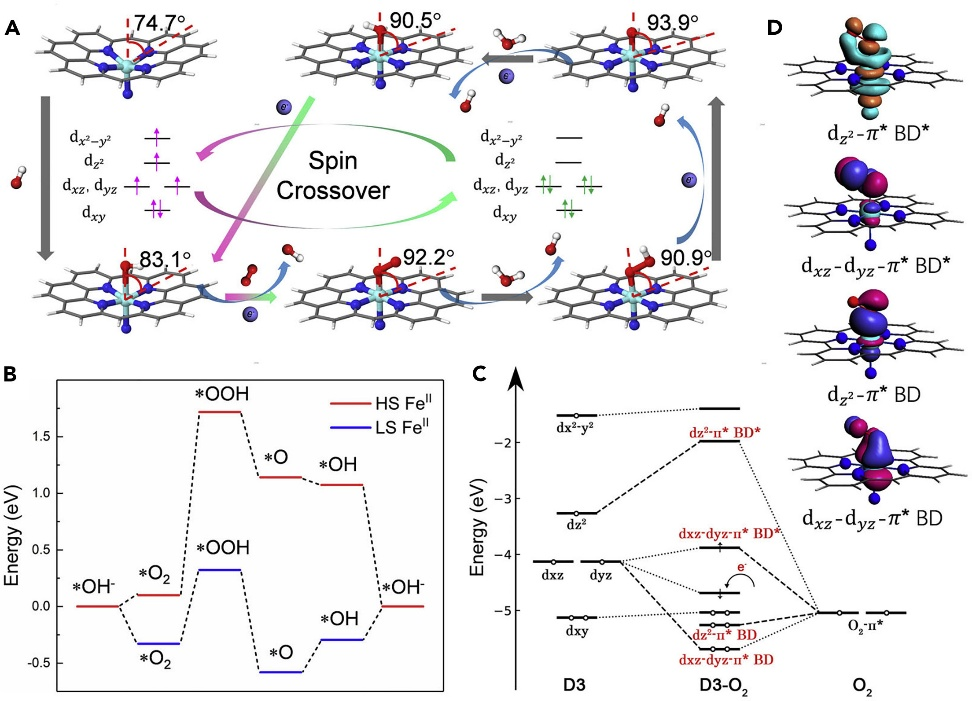
图9. (A)ORR过程中N-FeN₄C₁₀基团的结构与动力学特征,给出了优化后的几何构型(图中角度表示铁原子相对于N₄平面的偏离角度)。(B)N-FeN₄C₁₀基团在碱性介质中发生氧还原反应(U = 0.401 V)时的势能剖面。蓝色曲线表示伴随自旋交叉的情况,红色曲线表示无自旋交叉的情况。(C)O₂与N-FeN₄C₁₀基团之间的轨道相互作用示意图(主要相互作用发生在dxz、dyz轨道与O₂的π*轨道之间)。(D)O₂与N-FeN₄C₁₀基团相互作用的自然定域化分子轨道(NLMOs)示意图。